Help
Troubleshooting
I'm having trouble with the Java viewer displaying my job, what is wrong?
It is more than likely that you need to increase the amount of memory available to the java applet. The PDB has a good description on increasing the memory available to Java.
My question isn't answered here, how can I reach you?
Please use the contact form on our lab website
Tutorial
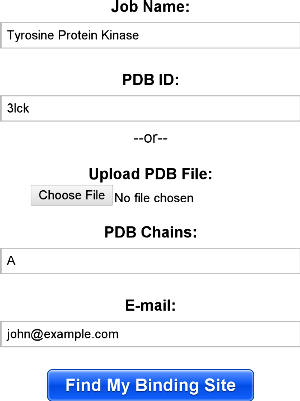
The interface for submitting jobs is show below. If you are using a structure from the PDB, you should specify the PDB id and chains, otherwise, you may upload your own PDB file. Note that any HETATM records are stripped out, removing any ligands in the file. Below, the user has decided to map chain a of the PDB id "3lck".
Upon submission of a job, you will see a success message as below. When the job is complete, you will receive an e-mail with a link to the results.

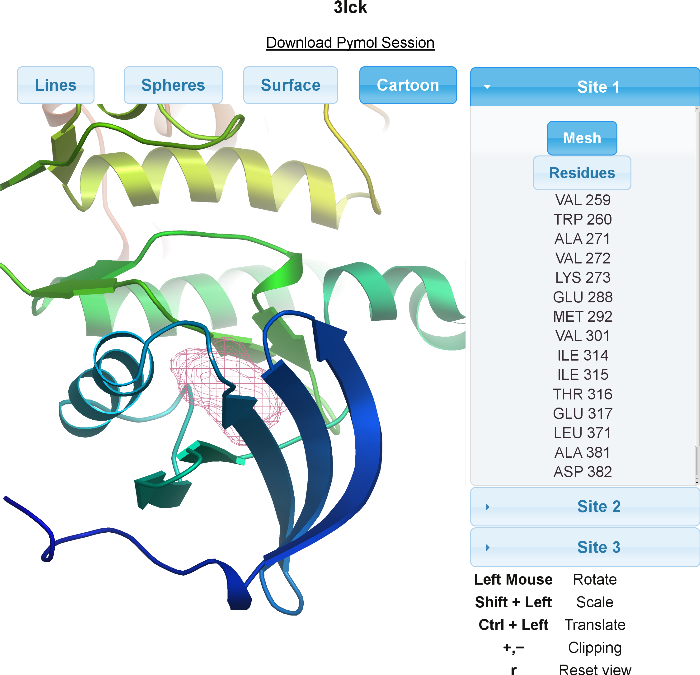
After receiving an e-mail with your results, you will have two ways of viewing the results. Firstly, the server will send a PyMOL session containing the protein, the detected sites, and the residues that surround the site. For those unfamiliar with PyMOL, a web interface for viewing the results is linked to in the e-mail. As shown below, the web interface contains a Java Applet. Mesh respresentations of the sites and sticks of the residues around the sites can be turned on and off along with various representations of the protein. Residues near each site are also enumerated for the user.